Lung Cancer- KRAS Mutation Panel
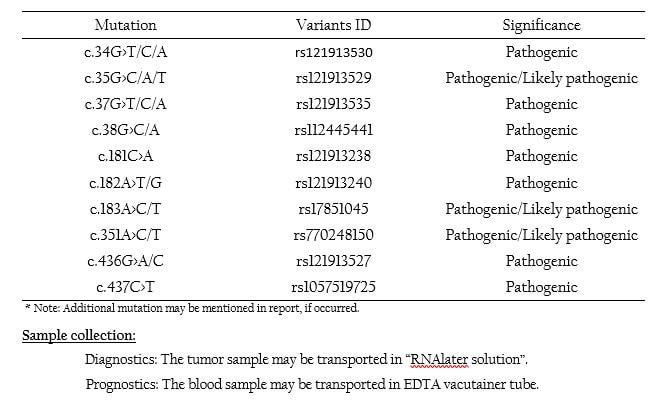
KRAS overview:
- The RAS genes are highly homologous but functionally distinct[1]
- RAS proteins are central mediators that downstream the growth factor receptor signaling and are critical for cell proliferation, survival, and differentiation.
- RAS can activate several downstream effectors, including the PI3K-AKT-mTOR pathway, which is involved in cell survival, and the RAS-RAF-MEK-ERK pathway, which is involved in cell proliferation.
- Activating mutations within the RAS gene result in constitutive activation of the RAS GTPase, even in the absence of growth factor signaling. The result is a sustained proliferation signal within the cell.
- Specific RAS genes are recurrently mutated in different malignancies. KRAS mutations are particularly common in colon cancer, lung cancer, and pancreatic cancer[2]
- KRAS and EGFR mutations are mutually exclusive[3]
- There are evidences that prove that somatic mutations of lung cancer harbours KRAS mutations[4]
- About 15 %to 25% of the Non-small cell lung carcinoma patients have KRAS mutations and almost 97% of the KRAS-mutant cases affect exon 2 and 3[5]
- The most common oncogenic mutation detected in patients with NSCLC is KRAS, which is found in 25% to 30% of lung adenocarcinomas, and associated with tobacco use[6&7]
- KRAS accounts for 90% of RAS mutations in lung adenocarcinomas, and approximately 97% of KRAS mutations in NSCLC involve codons 12 or 13[8]
- KRAS mutations are more common in lung cancers among smokers, while EGFR mutations are more frequent in never-smokers[9]
- KRAS mutations are more common in individuals with a history of cigarette use and are associated with resistance to EGFR-tyrosine-kinase inhibitors[10&11]
- About 30% of the KRAS mutations occur in lung adenocarcinomas and only 5% in squamous cell carcinoma subtype[12]
- These mutations impair the intrinsic GTPase activity of RAS and confer resistance to GTPase activators, thereby causing RAS to accumulate in its active GTP-bound state, which sustains the activation of RAS signaling[13]
- KRAS mutations in NSCLC tumor tissue are moderately associated with poor prognosis[14]
- The overall survival rate of NSCLC patients harbouring KRAS mutations is worse than patients with NSCLC and with wild-type KRAS[15]
- KRAS gene is prevalent in pancreatic carcinomas (>80%), colon carcinomas (40–50%), and lung carcinomas (30–50%), but are also present in biliary tract malignancies, endometrial cancer, cervical cancer, bladder cancer, liver cancer, myeloid leukemia[16] and breast cancer[17]
- NSCLCs, in which higher VEGF expression was observed in 50% of tumours bearing a KRAS gene mutation
- 50% of the NSCLC tumours shows higher expressions of vascular endothelial growth factor (VEGF) due to KRAS mutations[18]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. (2011). Nat Rev Cancer, 13:(11):761-74.
- Karnoub AE and Weinberg RA. (2008). Nat Rev Mol Cell Biol, 9(7):517-31.
- Kris MG. (2005). Oncologist. 10(2):23-29.
- Rodenhuis S, van de Wetering ML, Mooi WJ, et al. (1987). N Engl J Med, 317:929-935.
- Brose MS, Volpe P, Feldman M, et al. (2002). Cancer Res; 62:6997-7000.
- Barlesi F, Mazieres J, Merlio JP, et al. (2016). Lancet, 387(10026):1415-1426.
- Kris MG, Johnson BE, Berry LD, et al. (2014). JAMA, 311(19):1998-2006.
- Forbes S, Clements J, Dawson E, Bamford S, et al. (2006). Br J Cancer, 94:318-322.
- Shigematsu H, Lin L, Takahashi T, et al. (2005). J Natl Cancer Inst, 97(5):339-346.
- Le Calvez F. et al. (2005). Cancer Res. 65, 5076–5083.
- Pao W, et al. (2005). PLoS Med. 2:e17.
- Roberts PJ, Stinchcombe TE, Der CJ, et al. (2010). J Clin Oncol, 28:4769-77.
- Trahey M, McCormick FA. (1987). Science, 238:542-5.
- Aviel-Ronen S, Blackhall FH, Shepherd FA, et al. ((2006). Clin. Lung Cancer 8:30-38.
- Miyake M, Adachi M, Huang C, et al. (1999). Oncogene, 18:2397-404.
- Downward J. (2003). Biochemical Journal, 376: e9–e10.
- Schubbert S, Shannon K, and Bollag G, (2007). Nature Reviews Cancer, 7:4, 295-308.
- Konishi T, Huang CL, Adachi M, et al., (2000). International Journal of Oncology, 16: 3, 501-511.